Grouping and summarising data
2025-08-14
Counting and describing data
This notebook introduces two new functions: group_by()
and summarise(). The first function,
group_by(), is used to first tell a tibble/dataframe your
set of grouping variables, i.e., what do you want to group by. The
second function, summarise(), will allow you to ask for any
number of functions applied to your data based on that grouping
structure. To demonstrate, let’s look at some penguins!
Load tidyverse and the palmerpenguins data
set
obtaining mean values for different species of
penguins
We can calculate the average/mean of a variable using the base
function mean(). This function takes a vector of numbers or
a dataframe column and returns the average/mean. Lets use it to find the
average body mass of the penguins.
## [1] NAThere is at least one NA value in the dataframe, so the function is
returning NA. We can deal with this by using the
na.rm argument in mean. The default value of
na.rm is FALSE, meaning that NA
values will not be removed. Set the argument to TRUE in
order to remove the NA values from the calculations
## [1] 4201.754After removing the NA values, we see that the average
body mass of the penguins is about 4200 grams. However, in the notebook
using filter(), we developed a hypothesis that certain
species of penguins had higher body masses than others. Let’s confirm
this hypothesis by using mean() on three filtered versions
of the data.
## Adelie Chinstrap Gentoo
## 152 68 124In the line below, I wrap mean() around a
filter() call to penguins, which asks for just
the "Adelie" penguins. I also attach the $ to
the end of the filter function to call just the body_mass_g
column. I include na.rm = TRUE to filter out
NA.
So this line of code is saying “Give me the mean of the column
body_mass_g in the penguins data after
filtering all rows where species is equal to ‘Adelie’ and also remove
NA values during the calculation of the mean”. While this
is “efficient” in being a single line, it can be a bit difficult to take
in if you are unfamiliar with the different parts of the R syntax. (This
is a pedagogical decision, we will see later how group_by()
and summarise() improve on this.)
## [1] 3700.662We see that the average body mass of the Adelie penguins is lower than the average body mass of all the penguins. Let’s quickly look at the average body mass of the other two species:
## [1] 3733.088## [1] 5076.016It seems both the Adelie and Chinstrap penguins have a lower average
body mass than the entire population average, whereas the Gentoo
penguins are heavier. To understand this, we repeated the same thing
three times, which is usually a signal that we could be more efficient.
Moreover, we only see the means one-at-a-time. It would be far more
useful to obtain all of this information at the same time. This is where
group_by() and summarise() come in handy!
let’s group stuff!
The first thing we need to think about is what our grouping variable
will be. In this case, it should be relatively obvious that it is
species (this is the variable we used filter()
on three times). Knowing this, we can start to build
group_by() pipe.
It’s usually good to create a new tibble that contains your summary
data. Let’s create a new variable called bm_summary, which
is a copy of penguins. Then pipe into the
group_by() function. We want to group by
species, so add that into the group_by()
function.
Let’s start with the penguins data and pipe into
group_by(), like this:
Running this code doesn’t seem to do anything, but we have now create
a new tibble which contains metadata about a grouping structure imposed
on the data. You can verify this using glimpse(), which now
contains a Groups output.
## Rows: 344
## Columns: 8
## Groups: species [3]
## $ species <fct> Adelie, Adelie, Adelie, Adelie, Adelie, Adelie, Adel…
## $ island <fct> Torgersen, Torgersen, Torgersen, Torgersen, Torgerse…
## $ bill_length_mm <dbl> 39.1, 39.5, 40.3, NA, 36.7, 39.3, 38.9, 39.2, 34.1, …
## $ bill_depth_mm <dbl> 18.7, 17.4, 18.0, NA, 19.3, 20.6, 17.8, 19.6, 18.1, …
## $ flipper_length_mm <int> 181, 186, 195, NA, 193, 190, 181, 195, 193, 190, 186…
## $ body_mass_g <int> 3750, 3800, 3250, NA, 3450, 3650, 3625, 4675, 3475, …
## $ sex <fct> male, female, female, NA, female, male, female, male…
## $ year <int> 2007, 2007, 2007, 2007, 2007, 2007, 2007, 2007, 2007…You can also look at the tibble in the environment pane to see the grouping structure:
You can use the ungroup() function to remove this
grouping structure.
# The Group information is gone now.
bm_summary <- penguins %>%
group_by(species) %>%
ungroup() %>%
glimpse()## Rows: 344
## Columns: 8
## $ species <fct> Adelie, Adelie, Adelie, Adelie, Adelie, Adelie, Adel…
## $ island <fct> Torgersen, Torgersen, Torgersen, Torgersen, Torgerse…
## $ bill_length_mm <dbl> 39.1, 39.5, 40.3, NA, 36.7, 39.3, 38.9, 39.2, 34.1, …
## $ bill_depth_mm <dbl> 18.7, 17.4, 18.0, NA, 19.3, 20.6, 17.8, 19.6, 18.1, …
## $ flipper_length_mm <int> 181, 186, 195, NA, 193, 190, 181, 195, 193, 190, 186…
## $ body_mass_g <int> 3750, 3800, 3250, NA, 3450, 3650, 3625, 4675, 3475, …
## $ sex <fct> male, female, female, NA, female, male, female, male…
## $ year <int> 2007, 2007, 2007, 2007, 2007, 2007, 2007, 2007, 2007…let’s summarise stuff!
Okay, now that we can group, let’s use summarise() to
obtain the mean of each level of our grouping variable. To do so, pipe
into a summarise() function after the
group_by() function.
The summarise() function works similar to
mutate(). We create new values, so we first give our value
a new name. Call the new value mean_body_mass. We then set
the value to be the result of calling some sort of function on a column.
In this case, we want to call mean() on
body_mass_g. Don’t forget to include
na.rm = TRUE !
bm_summary <- penguins %>%
group_by(species) %>%
summarise(mean_body_mass = mean(body_mass_g, na.rm = TRUE)) %>%
glimpse()## Rows: 3
## Columns: 2
## $ species <fct> Adelie, Chinstrap, Gentoo
## $ mean_body_mass <dbl> 3700.662, 3733.088, 5076.016Notice what happens to the tibble, it’s now 3 rows long and 2 columns wide. If you look at the data in the data viewer, you see something like this:
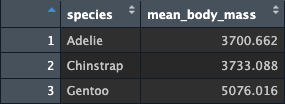
So the two remaining columns are (1) the column which was included in
group_by() and (2) the new variable that was created. We
have reduced (or summarised) our tibble into a smaller set of data. And,
moreover, we can clearly see the 3 means for the 3 species in one place.
Calling the name of the tibble prints this information straight to the
console for us, neat!
## # A tibble: 3 × 2
## species mean_body_mass
## <fct> <dbl>
## 1 Adelie 3701.
## 2 Chinstrap 3733.
## 3 Gentoo 5076.creating multiple values in summarise()
Just like mutate(), we can create as many variables as
we want within a single summarise() function. Let’s try
that now. When we report the mean/average of something, we usually also
report the standard deviation. You can obtain the standard deviation in
R using the sd() function. With this knowledge, let’s
create a new variable called sd_body_mass alongside the
mean. It will also require the na.rm = TRUE command
bm_summary <- penguins %>%
group_by(species) %>%
summarise(mean_body_mass = mean(body_mass_g, na.rm = TRUE),
sd_body_mass = sd(body_mass_g, na.rm = TRUE)) %>%
glimpse()## Rows: 3
## Columns: 3
## $ species <fct> Adelie, Chinstrap, Gentoo
## $ mean_body_mass <dbl> 3700.662, 3733.088, 5076.016
## $ sd_body_mass <dbl> 458.5661, 384.3351, 504.1162Just as before, there are 3 rows (one per level of species), but now we also have 3 columns. This should make sense, since we asked for a third value (standard deviation). Your data should look something like this:
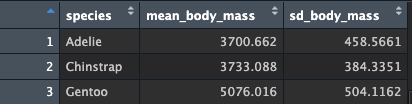
Keep going! Add the min() and max() values
to the data, all inside the same summarise() call. Give the
new variables the names min_body_mass and
max_body_mass:
bm_summary <- penguins %>%
group_by(species) %>%
summarise(mean_body_mass = mean(body_mass_g, na.rm = TRUE),
sd_body_mass = sd(body_mass_g, na.rm = TRUE),
min_body_mass = min(body_mass_g, na.rm = TRUE),
max_body_mass = max(body_mass_g, na.rm = TRUE)) %>%
glimpse()## Rows: 3
## Columns: 5
## $ species <fct> Adelie, Chinstrap, Gentoo
## $ mean_body_mass <dbl> 3700.662, 3733.088, 5076.016
## $ sd_body_mass <dbl> 458.5661, 384.3351, 504.1162
## $ min_body_mass <int> 2850, 2700, 3950
## $ max_body_mass <int> 4775, 4800, 6300Just like mutate, we can operate on variables we create
inside the summarise() function! So, after
creating the min/max body mass columns, we could then perform a
calculation to obtain the range() of the body mass (i.e.,
the distance between the minimum and maximum values). Instead of using a
function, let’s just subtract the max from the min.
bm_summary <- penguins %>%
group_by(species) %>%
summarise(mean_body_mass = mean(body_mass_g, na.rm = TRUE),
sd_body_mass = sd(body_mass_g, na.rm = TRUE),
min_body_mass = min(body_mass_g, na.rm = TRUE),
max_body_mass = max(body_mass_g, na.rm = TRUE),
range_body_mass = max_body_mass - min_body_mass) %>%
glimpse()## Rows: 3
## Columns: 6
## $ species <fct> Adelie, Chinstrap, Gentoo
## $ mean_body_mass <dbl> 3700.662, 3733.088, 5076.016
## $ sd_body_mass <dbl> 458.5661, 384.3351, 504.1162
## $ min_body_mass <int> 2850, 2700, 3950
## $ max_body_mass <int> 4775, 4800, 6300
## $ range_body_mass <int> 1925, 2100, 2350but how do you count things?
Pretty cool right - we can create as many values as we need in order
to describe our population. However, we are missing a pretty important
one…counting the members of our population! A shorthand you might see
for this in manuscripts and elsewhere is something like
n = x. There is happily a function called n()
which gives this exact information. Let’s try it out by asking for the
n of our penguins species. It’s as simple as adding
n = n(). I’m choosing to call it n, but we
could call it anything (right?)
bm_summary <- penguins %>%
group_by(species) %>%
summarise(n = n(),
mean_body_mass = mean(body_mass_g, na.rm = TRUE),
sd_body_mass = sd(body_mass_g, na.rm = TRUE),
min_body_mass = min(body_mass_g, na.rm = TRUE),
max_body_mass = max(body_mass_g, na.rm = TRUE),
range_body_mass = max_body_mass - min_body_mass) %>%
glimpse()## Rows: 3
## Columns: 7
## $ species <fct> Adelie, Chinstrap, Gentoo
## $ n <int> 152, 68, 124
## $ mean_body_mass <dbl> 3700.662, 3733.088, 5076.016
## $ sd_body_mass <dbl> 458.5661, 384.3351, 504.1162
## $ min_body_mass <int> 2850, 2700, 3950
## $ max_body_mass <int> 4775, 4800, 6300
## $ range_body_mass <int> 1925, 2100, 2350adding another grouping variable
The beauty of group_by() is that you can add any number
of grouping variables to make calculations with. So, right now the
summarise function has been operating across the three different types
of species in the data. but what if we wanted to also know this
information separated by year? It’s as simple as adding
year to the group_by call. Let’s try it out
and look at the population and mean body mass of penguins, by species,
by year:
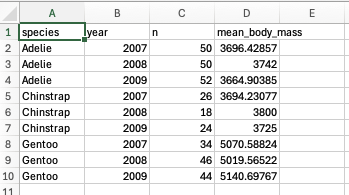
bm_year_summary <- penguins %>%
group_by(species, year) %>%
summarise(n = n(), mean_body_mass = mean(body_mass_g, na.rm = TRUE))## `summarise()` has grouped output by 'species'. You can override using the `.groups`
## argument.## # A tibble: 9 × 4
## # Groups: species [3]
## species year n mean_body_mass
## <fct> <int> <int> <dbl>
## 1 Adelie 2007 50 3696.
## 2 Adelie 2008 50 3742
## 3 Adelie 2009 52 3665.
## 4 Chinstrap 2007 26 3694.
## 5 Chinstrap 2008 18 3800
## 6 Chinstrap 2009 24 3725
## 7 Gentoo 2007 34 5071.
## 8 Gentoo 2008 46 5020.
## 9 Gentoo 2009 44 5141.adding a third grouping variable
Repeat the above, but also include the sex variable in
the summary. This has introduced a few rows where sex is NA. What is
going on? (Hint, look at the full data frame and find all the NA
values.)
bm_year_sex_summary <- penguins %>%
group_by(species, year, sex) %>%
summarise(n = n(), mean_body_mass = mean(body_mass_g, na.rm = TRUE))## `summarise()` has grouped output by 'species', 'year'. You can override using the
## `.groups` argument.## # A tibble: 22 × 5
## # Groups: species, year [9]
## species year sex n mean_body_mass
## <fct> <int> <fct> <int> <dbl>
## 1 Adelie 2007 female 22 3390.
## 2 Adelie 2007 male 22 4039.
## 3 Adelie 2007 <NA> 6 3540
## 4 Adelie 2008 female 25 3386
## 5 Adelie 2008 male 25 4098
## 6 Adelie 2009 female 26 3335.
## 7 Adelie 2009 male 26 3995.
## 8 Chinstrap 2007 female 13 3569.
## 9 Chinstrap 2007 male 13 3819.
## 10 Chinstrap 2008 female 9 3472.
## # ℹ 12 more rowsHow do i get this information outside of R?
You probably want to report this summary information in a manuscript,
likely even as a table. You could copy and paste this
information from R into somewhere else, but why do that when you could
easily output the information as a .csv file? We can do so with the
write_csv() function. The function takes two main arguments
- the name of the tibble you want to output, and the name you would like
to give the file. So, running the cell below will output the object
bm_year_summary and give it the name
'my_awesome_penguins_summary.csv'
If you don’t tell R where to save the file, it will be saved to
whatever the current working directory is. If you are using an R
markdown or notebook file, it should be the same directory of wherever
that file is saved. You can check the current working directory using
the getwd() function.
And if we were to open this up in a spreadsheet program, it might look like this: